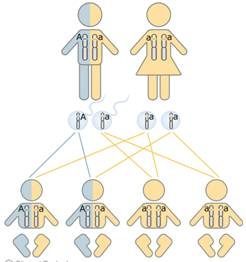
La
clonación(copia
idéntica de un organismo a partir de su ADN)
se puede definir como el proceso por el que se consiguen, de forma
asexual,
2 copias
idénticas de un organismo,
célula
o
molécula
ya
desarrollado.
Diferencias
entre Clonación Natural y Clonación Artificial:
Una
diferencia entre estas dos es que la primera es producida por la
propia
naturaleza, y la segunda es producto de la voluntad del hombre
a través de la manipulación del material genético a efectos de
crear un nuevo ser.
La
clonación Natural es un metodo de reproduccion asexual, donde la
fertilizacion no ocurre y solo hay un progenitor involucrado: se da
cuando una célula se divide, por el proceso de fisión,
proporcionando a la nueva célula los elementos metabólicos y
fisiológicos necesarios para permitirle su individualidad e
independencia.
En cambio, en la Clonación Artificial la fecundación se realiza
asistidamente de manera extracorpórea y atípica logrando engendrar
un ser asexualmente cuya principal caracteristica es tener un codigo
genético compartido con su genitor. Este tipo de clonacion consiste
en tomar un embrion de hasta 8 celulas y generar embriones identicos
preimplantatorios.
VENTAJAS DE LA VARIABILIDAD GENÉTICA:
La principal ventaja es el aumento de la capacidad de la supervivencia de la especie, ya que se podria decir que saca lo mejor del ADN de cada especie, para hacerse mas resistente a determinadas situaciones, osea que permite a una especie adaptarse a nuevos entornos y con ello diseminarse por ellos.
Los seres humanos utilizamos la variabilidad para crear razas y tambien variedades de maices, manzanas, calabazas, caballos, vacas, borregos, perros y gatos, entre otros.
Permite la evolución de las especies, además la existencia de mayor variabilidad genética implica mayor raza evolutiva.
FUNCIÓN VITAL QUE PERMITE LA VARIABILIDAD GENÉTICA:
La Meiosis es importante para la variabilidad genética, ya que gracias a ella existen diferencias de rasgos con respecto al padre y a la madre, y permite una mezcla. Además la Variabilidad permite la evolución de los organismos.
A través de la meiosis no solo se forman celulas haploides que haran posible la fecundacion y la reproduccion sexual, al mismo tiempo, como se ha visto, tambien se recombina, mediante entrecuzamiento cromosomico, la informacion genetica que pasa de una generacion a la siguiente, lo cual favorece la variabilidad genetica.
OPINIÓN GENERAL SOBRE LA CLONACIÓN ARTIFICIAL
Con respecto a la clonación artificial creemos que es muy útil siempre y cuando sepamos utilizarla correctamente. Si nos referimos al ámbito de la medicina es muy eficaz a la hora de clonar órganos, eso disminuiría la cantidad de donaciones.
Sin embargo, la idea de utilizarla para clonar seres humano, personas, creemos que es algo negativo.
En clonclucion, tiene ventajas y desventajas este facilitaria al ambito medico y es un gran avance para la ciencia; Por otro lado también tiene desventajas, como lo que lo clonado deja de ser natural, es decir, suplanta la naturaleza de la cosas.
Gregor Johann Mendel
Botánico austriaco .
Nació el 22 de julio de 1822, en Heinzendorf (hoy Hyncice, República Checa).
Hijo de un veterano de las guerras napoleónicas que explotaba una pequeña granja.
En
1841 su padre fue aplastado por el tronco de un árbol y se vio obligado
a vender sus propiedades. Su hermana le entregó su parte para ayudarle
en sus estudios eclesiásticos. Durante dos años estudió física y matemáticas en el Instituto Filosófico Olmütz. Ingresó en el monasterio de agustinos de Brünn (hoy Brno, República Checa) y a los veintiún años se convirtió en un novicio agustino y adoptó el nombre de Gregor.
Inició un curso de cuatro años de estudios en el Colegio Teológico de Brünn en 1845 y fue ordenado sacerdote en 1847.
Le asignaron el puesto de profesor delegado de matemáticas avanzadas en 1849. En el año 1850 suspende biología
en el examen de cualificación para el profesorado. Fue enviado a la
Universidad de Viena durante dos años para estudiar física práctica y
matemáticas, química, zoología, paleontología, botánica sistemática y
fisiología vegetal, que incluía las nuevas teorías celulares.
Pasado algún tiempo comenzó a trabajar como profesor suplente en la Escuela Técnica de Brünn
donde se dedicó de forma activa a investigar la variedad, herencia y
evolución de las plantas en un jardín del monasterio destinado a los
experimentos. Entre 1856 y 1863 cultivó y estudió al menos 28.000 plantas de guisante analizando con detalle siete pares de características de la semilla y la planta. Gracias a sus numerosos experimentos logró el enunciado de dos principios que más tarde serían conocidos como leyes de la herencia. Sus observaciones le llevaron también a acuñar dos términos que siguen empleándose en la genética de nuestros días: dominante y recesivo.
La llamada ley de la uniformidad de los híbridos
de la primera generación, dice que cuando se cruzan dos variedades de
individuos de razas puras ambos homocigotos para un determinado
carácter, todos lo híbridos de la primera generación son igual es fenotípicamente. Informó
de sus hallazgos en una reunión de la Sociedad para el estudio de la
Ciencias Naturales en Brno, y publicó sus resultados en las actas de
dicha sociedad, en el año de 1866. La importancia de sus hallazgos no
fue apreciada por otros biólogos de su época, y fueron despreciados por
espacio de 35 años. Sólo obtuvo el debido reconocimiento en 1900 por
parte de tres investigadores, uno de los cuales fue el botánico holandés
Hugo de Vries, y sólo a finales de la década de 1920 y comienzos
de 1930, se comprendió su verdadero alcance, en especial en lo que se
refiere a la teoría evolutiva. Sus experimentos posteriores con la vellosilla Hieracium,
no fueron concluyentes, y debido a la presión de otras ocupaciones, en
la década de 1870 había abandonado ya sus experimentos sobre la
herencia.
Gregor Mendel falleció el 6 de enero de 1884 en Brünn.
¿Qué dicen las 3 leyes de Mendel?
LEYES DE MENDEL
PRIMERA LEY DE MENDEL:
La primera ley de Mendel, también llamada: Ley de la uniformidad de
los híbridos de la primera generación, o simplemente Ley de la
Uniformidad. Esta ley dicta que, al cruzar dos variedades de una especie
de raza pura, cada uno de los híbridos de la primera generación tendrá
caracteres determinados similares en su fenotipo. Esto se debe a que las
razas puras tienen un gen dominante o un gen recesivo. El genotipo
dominante será entonces el que determine la característica o
características principales de la primera generación del cruce, pero al
mismo tiempo, también serán similares fenotípicamente entre sí, es
decir, entre cada individuo de la primera generación.
En el
experimento realizado por Mendel para obtener la primera de las leyes de
Mendel, utilizaba una especie de chícharos que producían semillas
amarillas como gen dominante y otra que tenía un gen recesivo que
producía semillas verdes, por lo tanto, el alelo que llamaremos “A” daba
el color amarillo por encima del alelo “a” que producía el color verde.
El producto del cruce eran plantas que
producían semillas amarillas.
Sigue leyendo aquí para aprender más sobre la Ley De Mendel.
SEGUNDA LEY DE MENDEL:
La segunda ley de Mendel, también conocida como la Ley de la
Segregación, Ley de la Separación Equitativa, o hasta Ley de Disyunción
de los Alelos. Esta dictamina que para que exista la reproducción de
dos individuos de una especie, primero debe existir la separación del
alelo de cada uno de los pares para que de esta manera se transfiera la
información genética al hijo. Un alelo es, la variante genética que
permite determinar un rasgo o carácter. Existen entonces, alelos
dominantes y alelos recesivos.
Por
esto, es que la segunda de las leyes de Mendel se la llama como de
segregación o separación, ya que cada padre, aporta un alelo que se
separa de cada uno, para formar un individuo en una nueva generación.
Sigue leyendo aquí para aprender más sobre la Segunda Ley De Mendel.
Mendel, en su experimento, obtuvo solo semillas amarillas en la primera
generación, pero en la segunda generación, los alelos se separaron para
formar nuevas semillas verdes en menor proporción que las amarillas,
pero aun así existentes. Esta sería la proporción:
TERCERA LEY DE MENDEL:
Tercera Ley De Mendel
La tercera ley de Mendel, también
llamada Ley de la Herencia Independiente de Caracteres o Ley de la
Asociación Independiente. Según Mendel, hay rasgos heredados que se
obtienen de forma independiente, sin relación con el fenotipo, lo cual
no afecta al patrón de herencia de otros rasgos. Esta ley se cumple en
los genes que no están ligados, es decir que se encuentran en diferentes
cromosomas o que están en zonas muy separadas del mismo cromosoma.
Mendel,
para concluir la tercera de las leyes de Mendel, realizó un cruce de
plantas de chícharos que producían semillas amarillas y llanas, con
chícharos que producían semillas verdes y con textura irregular. Estas
eran homocigóticas para los dos caracteres de textura y color. Se
concluía que la ley de uniformidad estaba presente, pues con la primera
generación se pudo obtener semillas amarillas y lisas.
Sin
embargo, al cruzar esta primera generación para obtener una segunda
generación, se observan nuevos tipos de semillas con caracteres diversos
pero relacionados con la generación parental, se obtuvieron semillas
amarillas y lisas, amarillas y rugosas, verdes y lisas, y verdes y
rugosas. Sigue leyendo aquí para aprender más acerca de Tercera Ley De Mendel.
Leyes De Mendel Resumidas
Para
terminar con las Leyes De Mendel Resumidas, se puede decir que, la
primera Ley de Mendel dice que si se cruzan dos padres de raza pura con
diferentes rasgos, la primera generación tendrá similitudes entre sí y
guardará un carácter del padre con el alelo dominante. La segunda ley
dice que, los factores genéticos se separan de cada uno de los padres en
alelos individuales que se juntarán para procrear una descendencia con
las características de la primera generación, pero en la segunda
generación, se manifiestan nuevos rasgos genéticos observados en los
padres pero unidos de manera aleatoria en la descendencia de la primera
generación. Y la tercera ley de Mendel dice que, además existen rasgos
generados de forma independiente, a través de cromosomas alejados que no
intervienen entre sí, y al igual que en la segunda ley, esta tercera de
las leyes de Mendel se manifiesta con más claridad en la segunda
generación de individuos.
DEFINICIONES:
Fenotipo:
Se entiende por fenotipo todos aquellos rasgos particulares y
genéticamente heredados de cualquier organismo que lo hacen único e
irrepetible en su clase. El fenotipo se refiere principalmente a
elementos físicos y morfológicos tales como el color de cabello, el tipo
de piel, el color de ojos, etc., pero además de los rasgos que hacen al
desarrollo físico también incluye a aquellos asociados al
comportamiento y a determinadas actitudes.
Genotipo: a instancias de la Biología, el genotipo resulta ser el conjunto de
genes característicos de cada especie, vegetal o animal, es decir, el
genotipo son los genes en formato de ADN que un animal, un vegetal o un
ser humano recibe de herencia de parte de sus dos progenitores, madre y
padre, y que por tanto se encuentra conformado por las dos dotaciones de
cromosomas que contienen la información genética del ser en cuestión.
Dominante: En genética este termino se utiliza para definir al carácter que se impone sobre todo y aparece en el organismo. Dando lugar a que el fenotipo heredado se exprese como tanto si esta presente en los dos cromosomas del par (en cuyo caso seria homocigótico) como si solo esta presente en uno (Heterocigoótico)
Recesivo:
Carácter heredado que esta imposibilitado de manifestarse ya que hay un
carácter dominante presente, para que este pueda manifestarse el
organismo debe poseer dos copias del mismo pero una provenientemente del
padre y una provenientemente de la madre.
Estos dos últimos conceptos en general son relativo debido a que pueden
existir mas de dos variantes de un gen distinto. Entonces en este caso
podría ser recesivo con respecto al segundo.
Monohibridismo: son los cruzamientos entre dos seres vivos de una
misma especie, que difieren en un solo rasgo. También es el estudio de
un solo rasgo en los cruzamientos de individuos distintos. Gregor
Mendelhizo cruzamientos monohibridicos para comprobar su primera ley.
Dihibridismo:El se produce al cruzase dos individuos que se diferencian en dos características. En el cruzamiento entre dos lineas puras o naturales que se diferencien en 2 caracteres o de los que estudiaron solamente estas 2 diferencial.
Codominancia:Se denomina codominancia al proceso por el cual un individuo manifiesta
dos características genéticas dominantes.
Estado en que un gen expresa su característica en el heterocigoto de
modo equivalente a su par. Los alelos del gen se expresan al mismo
tiempo dando origen al gameto masculino que después se unira al
femenino. en pocas palabras la codominancia o dominancia compartida es
en la cual los caracteres dominantes dan una característica fenotípica
tanto del padre como de la madre a la siguiente generación.
La genética humana describe el estudio de la herencia biológica en los seres humanos.
El estudio de la genética humana puede ser útil ya que puede responder preguntas sobre la naturaleza humana, comprender el desarrollo eficaz para el tratamiento de enfermedades en la genética de la vida humana.
Métodos de estudio de la herencia humana
Citogenéticas: estudio cariotipo humano para detectar mutaciones cromosómicas y genómicas. Método doble: consiste en el estudio de la herencia humana en el estudio de los rasgos en los gemelos.
Herencia ligada al sexo
Herencia transmitida por cromosomas sexuales. Este tipo de herencia se transmite de manera desigual en la descendencia segun el sexo.
Un ejemplo de este tipo de herencia es la que determina la transmisión del daltonismo y la hemofilia.
Daltonismo
Defecto de la vista que consiste en no distinguir ciertos colores o confundirlos con otros.
Síntomas: -Dificultad para ver los colores y sus brillos en forma casual.
-Incapacidad de establecer una diferencia entre los diferentes tonos de un color u otros similares.
Causas: Esta alteración tiene un origen genético, se trata de un trastorno de herencia ligado al sexo es decir, el gen afectado se encuentra en uno de los cromosomas sexuales.
Tratamientos: No existe un tratamiento para el daltonismo congénita por lo general, el daltonismo no causa una discapacidad importante.
Hay lentes de contacto y anteojos especiales que pueden ayudar.
Hemofilia
Es una enfermedad genética recesiva que impide la buena coagulación de la sangre.
Está relacionada con el cromosoma X.
Síntomas: Sangrado prolongado. Las hemorragias más graves son las que se producen en:
Articulaciones.
Cerebro.
Ojo.
Lengua.
Garganta.
Riñones.
Hemorragias digestivas, genitales, nasales.
Causas: Es hereditaria, es decir que ésta afección puede transmitirse de padres a hijos a través de los genes.
En pocas ocasiones se presenta como trastornos adquiridos.
Tratamientos: En la actualidad no hay ningún tratamiento curativo disponible y lo único que se puede hacer es corregir la tenencia hemorrágica administrando por una vía intravenosa el factor de coagulación que falta.
Como posibles tratamientos existen los caseros:
El reposo corporal.
Colocar hielo sobre la zona afectada.
jueves, 3 de noviembre de 2016
Teoría cromosómica de Sutton y Boveri
Teoría cromosómica de la herencia. Más conocida como Teoría cromosómica de Sutton y Boveri, enuncia que los alelos mendelianos están localizados en los cromosomas. Esta teoría fue desarrollada independientemente en 1902 por Theodor Boveri y Walter Sutton . Permaneció en controversia hasta 1915, cuando Thomas Hunt Morgan consiguió que fuera aceptada por el mundo.
Fundamentos
Los rasgos de un nuevo individuo son determinados por genes específicos presentes cromosomas heredados del padre y la madre. Los humanos tienen 100.000 genes aproximadamente en los 46 cromosomas.
Los genes que se localizan en el mismo cromosoma tienden a ser
heredados juntos y por esta razón se conoce como genes ligados. En las células
somáticas, los cromosomas se presentan como 23 pares homólogos para
formar el número diploide de 46. Hay 22 pares de cromosomas apareados,
los autosomas y un par de cromosomas sexuales.
La teoría cromosómica de Sutton y Boveri enuncia que los alelos mendelianos están localizados en los cromosomas.
Esta teoría fue desarrollada independientemente en 1902 por Theodor Boveri y Walter Sutton. También se denomina, a veces, teoría cromosómica de la herencia.
La teoría permaneció controvertida hasta 1915, cuando Thomas Hunt Morgan consiguió que fuera universalmente aceptada después de sus estudios realizados en Drosophila melanogaster.
Si el par de cromosomas sexuales es xx el individuo es genéticamente femenino, si el par es xy, el individuo es genéticamente masculino.
Un cromosoma de cada par proviene del gameto materno, el ovocito, y el otro componente del par proviene del gameto paterno, el espermatozoide. Así, cada gameto contiene un número haploide de 23 cromosomas y la unión de los gametos en la fecundación restaura el número diploide de 46.
Las ideas fueron:
La teoría celular, que concibe la célula como la unidad viva autónoma más pequeña; según esta idea, las células son las unidades fundamentales tanto de los organismos unicelulares (como bacterias o protistas), como de los multicelulares, y además constituyen los vehículos de propagación de losorganismos vivos: las esporas, el esperma y los huevos. Un punto muy importante es que la teoría celular considera que las nuevas células proceden de las antiguas, como lo expresó Rudolf Virchow: 'Omnis cellula ex cellula'.
El paradigma mendeliano: según las ideas de Gregor Mendel los elementos heredables se basan en unidades, partículas individuales que pasan de una generación a la siguiente mediante los mecanismos reproductivos. Estas unidades existen en parejas (los alelos), para los cuales pueden existir diferentes variantes. Estas variantes coexisten en los híbridos, y pasan a la generación siguiente en forma de copia simple. Es decir, Mendel establecía el comportamiento combinatorio de los genes (a los que él denominó 'factores') y su segregación en la descendencia.
Los estudios sobre fertilización realizados por Edouard Van Beneden en 1883 demostraron que los pronúcleos masculino y femenino en el zigoto de Ascaris contribuyen cada uno con un juego de cromosomas a la primera división celular. Por otro lado, un influyente citólogo de la época, August Weismann, propuso la cromatina como material hereditario. Incluso estableció la teoría de la línea germinal, y predijo algún tipo de reducción de la información antes de la formación de los gametos. Sin embargo Weismann, en su teoría cromosómica de la herencia de 1892, consideraba que cada cromosoma mitótico contenía el genoma completo de la línea germinal.
Mitosis
Es el proceso por medio del cual una célula se proviene, dando origen a dos células hija que son genéticamente idénticas a la célula madre. Cada célula hija recibe el complemento total de 46 cromosomas.
Antes de que una célula entre en mitosis
cada cromosoma replica su ácido desoxirribonucleico. Durante esta fase
de replicación los cromosomas son extremadamente largos, están dispersos
en forma difusa en el núcleo y no pueden ser reconocidos con el microscopio óptico.
Al comienzo de la mitosis, los cromosomas empiezan a enroscarse, a
contraerse y condensarse; estos eventos señalan el principio de la
profase. Cada cromosoma consiste ahora en 2 unidades paralelas, las
cromátidas, que se encuentran unidas en una región estrecha común a
ambas denominada centrómetro.
Durante la profase, los cromosomas continúan condensándose y se
vuelven más cortos y gruesos, pero solo en la prometafase se pueden
distinguir las cromáticas. En el curso de la metafase los cromosomas se
alinean en el plano ecuatorial y entonces resulta claramente visible su
estructura doble.
Cada cromosoma está unido por microtúbulos que se extienden desde
el centrómero hasta el centríolo, formando el uso mitótico. Poco
después, el centrómetro de cada uno de los cromosomas se divide,
señalando el comienzo de la anafase, seguida por la migración de las
cromátidas hacia los polos opuestos del uso.
Por último, durante la telofase los cromosomas se desarrollan y alargan, la envoltura nuclear se reconstituye y el citoplasma se divide. Cada célula hija recibe la mitad del material cromosómico duplicado.
Meiosis
Es la división celular que se produce en la célula germinales para generar los gametos femeninos y masculinos. En esta se efectúan 2 divisiones celulares sucesivas, la meiosis I y la meiosis II que reducen el número de cromosomas a un número haploide de 23.
Las células germinales replican su DNA al comienzo de la primera
división meiótica, de forma tal que cada uno de los 46 cromosomas se
duplica y queda constituido por 2 cromátidas hermanas. Pero, a
diferencia de lo que ocurre en la mitosis, los cromosomas
homólogos se aparean alineados entre si mediante un proceso denominado.
El apareamiento es exacto y punto a punto, excepto para la combinación
de X-Y. Luego, los homólogos apareados se separan quedando uno para cada
uno de las 2 células hijas. Poco tiempo después la meiosis II separa
las cromátidas hermanas. Finalmente cada gameto contiene 23 cromosomas.
Entrecruzamiento
Son eventos críticos que se producen durante la meiosis I y consisten en el intercambio de segmentos de cromátidas entre cromosomas
homólogos apareados. Los segmentos de cromátidas se rompen y son
intercambiados entre cromosomas homólogos separados. Durante la
separación de los cromosomas homólogos, los sitios de intercambio
permanecen transitoriamente unidos y la estructura cromosómica tiene en
estas circunstancias a la letra X y se denomina quiasma.
En cada meiosis I se producen de 30 a 40 entrecruzamientos, que son más frecuentes entre los genes localizados distantes entre sí en un cromosoma.
Fuentes
Colectivo de autores et Langman. Embriología médica.
Los estudios de Theodor Boveri
Pesar de que en tiempos de Theodor Boveri se aceptaba que una célula procede de la https://www.blogger.com/blogger.g?blogID=8471508263891909404#editor/target=post;postID=5855798638641481125;onPublishedMenu=overview;onClosedMenu=overview;postNum=2;src=postnamedivisión binaria de una célula madre, no estaba claro cómo la cromatina (que August Weismann
denominaba el “plasma germinal”) presente en el núcleo se transmite a
las células hijas de manera que ambas son idénticas a la célula
original, tras la “metamorfosis nuclear” observada por Walther Flemming, en la cual la masa nuclear se transforma en hebras definidas (los cromosomas)
que se mueven en el interior celular y luego vuelve a su estado
original. A pesar de que se suponía que dichas hebras transportaban el
material hereditario, el mecanismo permanecía desconocido, hasta que
Boveri demostró que los cromosomas son orgánulos permanentes que se
condensan durante la mitosis y permanecen difusos durante la interfase.
Además de establecer la individualidad y la permanencia de los
cromosomas, Boveri dio una descripción moderna del aparato mitótico,
pues fue el primero en identificar los centrosomas y definir el papel del huso mitótico
en la distribución de los cromosomas en los polos opuestos de la célula
madre, que darán lugar a las células hijas. Los trabajos de Boveri en Ascaris
y en embriones de erizos de mar le permitieron observar divisiones
celulares defectuosas, como mitosis multipolares, mitosis monopolares o
medios husos, que después fue capaz de inducir experimentalmente. Esto
le permitió definir tres reglas(1888,1904):
Los cromosomas durante mitosis son dobles (presentan dos cromátidas),
y cada parte presenta un lateral que se enfrenta hacia un polo del
huso; esta regla implica la idea de que un cromosoma sólo puede
dividirse entre dos células hijas, y la presencia de los cinetocoros, aún no descubiertos, enfrentados en las dos cromátidas para el anclaje de los microtúbulos. Boveri distinguió incluso dos tipos de cromosomas, los que tienen un centrómero localizado (en el erizo de mar) y los que lo tienen difuso (Ascaris).
Los cromosomas están conectados a ambos polos del huso a través de microtúbulos (MTs).
Cada cromátida está unida a uno de los dos polos y sólo a uno.
Boveri también identificó que las cromátidas se duplican durante la interfase (1904), y dedujo una correlación muy precisa entre el número cromosómico (la cantidad de cromatina) y el tamaño del núcleo (1905).
De esta forma, para el ciclo cromosómico, Boveri estableció tres
sucesos clave: duplicación de la cromatina durante el periodo de reposo
(interfase), la individualización de las cromátidas durante la
condensación cromosómica y la distribución de las cromátidas en anafase,
una descripción que encaja perfectamente con la visión actual de los
eventos cromosómicos durante el ciclo celular.
también describió el centrosoma por vez primera en Ascaris en 1887,
definiéndolo como un "orgánulo especializado en la división celular".
Boveri identificó claramente el centrosoma como un par de centriolos
rodeados por un material especial, capaz de ensamblar una "esfera de
arquiplasma" que contiene todos esos elementos, que a su vez generan de
forma transitoria una "astrosfera". En 1900, Boveri estableció que los
centrosomas son orgánulos celulares de una única copia. A través de su observación de la dinámica de los cromosomas, llegó a la conclusión de que un huso mitótico
bipolar típico consiste en realidad de dos medios husos, cada uno
generado por un centrosoma, que se mantienen unidos por el conjunto de
los cromosomas dobles unidos en el extremo de cada áster, de tal manera
que cada cromosoma está unido a ambos polos, y sólo a uno por cromátida.
Por tanto, dedujo que durante la formación de la placa metafásica,
existen fuerzas cromosómicas que parecen contrarrestar la repulsión
existente entre los áster.
Sin embargo, dado que los cromosomas de la línea germinal de Ascaris
son estructuralmene muy polimórficos, Boveri no pudo distinguir la
presencia de cromosomas homólogos en Parascaris equorum, ni en mitosis ni en el estado de sinapsis meiótica. Fue Walter Sutton quien reconoció la presencia de cromosomas individuales, identificables
por su tamaño, en espermatocitos de saltamontes, y demostró que 2
cromosomas similares siempre se aparean durante la meiosis.
Sin embargo, el hecho de que existan cromosomas morfológicamente
distintos no excluye que contengan información genética similar. En
1902, Boveri excluyó esta posibilidad mediante un ingenioso análisis de
dispermia (fertilización por más de un espermatozoide) en erizos de mar,
demostrando que los cromosomas no son equivalentes en el desarrollo
embrionario.
Mediante un análisis cuantitativo publicado en 1907, Boveri estimó el
número de "genóforos" que son esenciales para controlar la ontogenia,1 que corresponde al número haploide de cromosomas.
A partir de los datos de todos sus estudios citogenéticos, Boveri
llegó a la conclusión de que el aparato meiótico no distingue los
cromosomas homólogos en función de su origen paterno o materno. Por
tanto, esta división puede generar combinaciones múltiples de cromosomas
(2") para crear nuevos juegos haploides en los gametos.
Boveri resumió sus estudios en sus "Results on the constitution of the chromatic substance in the cell nucleus"
(Resultados sobre la constitución de la sustancia cromática del núcleo
celular), en el que combinaba los hechos observados sobre los cromosomas
con las leyes Mendelianas de la herencia. Como él mismo expresó:"Vemos que dos áreas de estudio que se han desarrollado de manera
independiente han producido resultados que son tan armoniosos como si
uno los hubiera derivado teóricamente del otro".
La confirmación de Thomas Morgan
Thomas Hunt Morgan tenía una formación de biólogo del desarrollo, habiendo recibido un Ph.D. en 1890 en la Universidad Johns Hopkins por sus estudios en el desarrollo de las arañas marinas, un grupo especializado de invertebrados,
y en 1891 aceptó un puesto de enseñanza en el Bryn Mawr College. En
1904 la Universidad de Columbia anunció la creación de un nuevo puesto
en zoología experimental, y se lo ofreció a Morgan, quien era amigo de
largo tiempo del director del departamento de zoología, E.B. Wilson.
Wilson convenció a Morgan de que la clave para entender el desarrollo
(esto es, como una célula, el huevo, genera un individuo completo) era
entender la herencia, ya que éste es el medio a través del cual el óvulo
y el espermatozoide transmiten las características de los individuos de
generación en generación.
Morgan inició sus estudios en ratas y ratones, pero éstos se
reproducen tan despacio que no resultaban convenientes para hacer
estudios sobre herencia. Buscando un organismo más apropiado, se decidió
por Drosophila melanogaster,
la mosca de la fruta, debido a sus características: es un organismo
pequeño (3 mm), fácil de mantener en el laboratorio (se pueden recoger
un millar en una botella de cuarto de litro), es fértil todo el año y
muy prolífica (produce una generación cada 12 días, o 30 generaciones al
año). Además los machos y las hembras se distinguen con facilidad, y el
desarrollo embrionario ocurre en el exterior, lo que facilita el
estudio de las mutaciones en el desarrollo. Por último, Drosophila
tiene sólo 4 pares de cromosomas, todo lo cual le convierte en un
organismo muy apropiado para los estudios sobre herencia. Los estudios
de Morgan con Drosophila comenzaron en 1907. Inicialmente, su
intención era mantener varias generaciones, esperando que apareciera un
mutante ocasional, algo que Hugo de Vries acababa de observar en plantas. Sin embargo, después de dos años manteniendo las moscas, sus esfuerzos permanecían vanos.
que los genes deben residir en los cromosomas
que cada gen debe residir en un cromosoma concreto
y que el carácter "color de ojos" debe residir en el cromosoma X y
estar ausente en el cromosoma Y, siendo el rojo el color dominante.
Posteriormente, Morgan razonó que los cromosomas son ensamblajes de
genes, puesto que caracteres que se encuentran en un cromosoma
determinado tienden a segregar juntos. Sin embargo, Morgan observó que
esos caracteres "ligados" en ocasiones se separan. A partir de aquí,
Morgan dedujo el concepto de recombinación
de cromosomas: postuló que dos cromosomas apareados pueden intercambiar
información, e incluso propuso que la frecuencia de recombinación
depende de la distancia entre ambos. Cuanto más cerca estén dos genes en
un cromosoma, mayor será la probabilidad de que se hereden juntos, y
cuanto mayor sea la distancia entre ellos, mayor será la probabilidad de
que se separen debido al proceso de entrecruzamiento (crossing-over).
En resumen, Morgan sugirió que la intensidad del ligamiento entre dos
genes depende de la distancia entre ellos en un cromosoma. Basándose en
esas observaciones, un estudiante del grupo de Morgan, Alfred Henry Sturtevant,
llegó a la conclusión de que las variaciones en la intensidad de
ligamiento podían utilizarse para mapear los genes en los cromosomas,
definiendo la distancia relativa unos de otros: un año después de que
Morgan hubiera identificado la mosca de ojos blancos, Sturtevant
estableció el mapa genético para los genes ligados al sexo. Hoy en día,
el Morgan es la unidad de medida de las distancias a lo largo de los
cromosomas en la mosca, el ratón y en humanos.
Morgan fue galardonado con el Premio Nobel de Fisiología o Medicina en 1933 por la demostración de que los cromosomas son portadores de los genes.
jueves, 20 de octubre de 2016
ADN RECOMBINANTE
-¿Qué es?
El ADN recombinante es resultado del uso de diversas técnicas que los biólogos moleculares utilizan para manipular las moléculas de ADN, y difiere de la recombinación genética que ocurre sin intervención dentro de la célula. -¿Cuál es su función?
El proceso consiste en tomar una molécula de ADN de un organismo, sea un virus, planta o una bacteria y en el laboratorio manipular y ponerla de nuevo dentro de otro organismo. -Aplicaciones médicas y otros.
Permite la posibilidad de utilizar plantas y alimentos transgénicos, así como microorganismos modificados genéticamente para producir fármacos y otros productos de utilidad para el hombre, entre los que se pueden citar: la insulina humana, la hormona del crecimiento, interferones, la obtención de nuevas vacunas o del ADN recombinante se ha logrado obtener plantas transgénicas resistentes a insectos, hongos, bacterias y herbicidas, con mayores características de calidad durante la pos cosecha y con alto contenido nutricional.
También ha permitido la clonación, expresión y producción mediante esta técnica de diversos antígenos, por ejemplo: la vacuna contra la hepatitis B, y la vacuna del virus del papiloma humano (HPV)
-Pros y contras. Pros: La tecnología de recombinación del ADN se utiliza para producir hormonas para mujeres con problemas de fertilidad y los anteriormente nombrados en aplicaciones. Contras: Para la salud, existen riesgos de transferir toxinas de una forma de vida a otra, lo que podría dar lugar a reacciones alérgicas imprevistas.
Existe el riesgo de que bacterias y virus modificados escapen de los laboratorios de alta seguridad e infecten a la población humana o animal.
-Noticia
1) Título: La vacuna contra el zika ya funciona en monos.
Una de las vacunas que se utilizó en los monos fue diseñada a partir del ADN recombinante, que está comenzando a ser utilizada. Este método que también se está utilizando en el desarrollo de la vacuna del bolla, consiste en aislar una molécula de ADN del virus que se desea combatir para introducirlo en el ADN de otro mosquito.
-www.elespanol.com
2) Título: Lanzaron la primera vacuna clostridial recombinante para bovinos y ovinos.
Se llama clostrimiq REC, producto elaborado con el soporte de la biología molecular.
La vacuna clostrimiq evita distraer el sistema inmune con otros antígenos asociaos como diferentes toxinas y bacterianos que generan una respuesta inmune que no protege contra la enfermedad y se incorpora sin que cumpla una función protectora.
-economía.tierra. com.ar
PROYECTO GENOMA HUMANO
El
proyecto, dotado con 3000 millones de dólares, fue fundado en 1990 en
el Departamento de Energía y los Institutos Nacionales de la Salud de
los Estados Unidos, bajo la dirección del doctor Francis Collins, quien
lideraba el grupo de investigación público, conformado por múltiples
científicos de diferentes países, con un plazo de realización de 15
años. Debido a la amplia colaboración internacional, a los avances en el
campo de la genómica, así como los avances en la tecnología
computacional, un borrador inicial del genoma fue terminado en el año
2000 (anunciado conjuntamente por el expresidente Bill Clinton y el
ex-primer ministro británico Tony Blair el 26 de junio de 2000),
finalmente el genoma completo fue presentado en abril del 2003, dos años
antes de lo esperado. Un proyecto paralelo se realizó fuera del
gobierno por parte de la Corporación Celera. La mayoría de la
secuenciación se realizó en las universidades y centros de investigación
de los Estados Unidos, Canadá, Nueva Zelanda, Gran Bretaña y España.
El
genoma humano es la secuencia de ADN de un ser humano. Está dividido en
fragmentos que conforman los 23 pares de cromosomas distintos de la
especie humana (22 pares de autosomas y 1 par de cromosomas sexuales).
El genoma humano está compuesto por aproximadamente entre 22500 y 25000
genes distintos. Cada uno de estos genes contiene codificada la
información necesaria para la síntesis de una o varias proteínas (o ARN
funcionales, en el caso de los genes ARN). El "genoma" de cualquier
persona (a excepción de los gemelos idénticos y los organismos clonados)
es único.
Conocer la secuencia completa del genoma
humano puede tener mucha relevancia cuanto a los estudios de biomedicina
y genética clínica, desarrollando el conocimiento de enfermedades poco
estudiadas, nuevas medicinas y diagnósticos más fiables y rápidos. Sin
embargo descubrir toda la secuencia génica de un organismo no nos
permite conocer su fenotipo. Como consecuencia, la ciencia de la
genómica no podría hacerse cargo en la actualidad de todos los problemas
éticos y sociales que ya están empezando a ser debatidos. Por eso el
PGH necesita una regulación legislativa basada en la ética.
Antes
de los ochenta ya se conocía la secuencia de genes sueltos de algunos
organismos, como también se conocían los genomas de entidades
subcelulares, tales como virus y plásmidos. Así pues, no fue hasta 1986
cuando el Ministerio de Energía (DOE), concretó institucionalmente el
Proyecto Genoma Humano (PGH) durante un congreso en Santa Fe. El PGH
contaba con una buena suma económica y sería utilizado para estudiar los
posibles efectos de las radiaciones sobre el ADN. Al siguiente año, en
el congreso de biólogos en el Laboratorio de Cold Spring Harbor, el
Instituto Nacional de la Salud (NIH) quiso participar del proyecto al
ser otro organismo público con mucha más experiencia biológica, si bien
no tanta en la organización de proyectos de esta magnitud.
El
debate público que suscitó la idea captó la atención de los
responsables políticos, no solo porque el Proyecto Genoma Humano era un
gran reto tecnocientífico, sino por las tecnologías de vanguardia que
surgirían, así como porque el conocimiento obtenido aseguraría la
superioridad tecnológica y comercial del país. Antes de dar luz verde a
la iniciativa del PGH se necesitó por un lado el informe de 1988 de la
Oficina de Evaluación Tecnológica del Congreso (OTA) y el del Consejo
Nacional de Investigación (NRC). Ese año se inauguró HUGO (Organización
del Genoma Humano) y James D. Watson fue nombrado alto cargo del
proyecto. Sería reemplazado por Francis Collins en abril de 1993, en
gran parte por su enemistad con Bernadine Healy que era su jefe por
aquel entonces. Tras esto el nombre del Centro cambió a Instituto
Nacional de Investigaciones del Genoma Humano (NHGRI).
En
1990 se inauguró definitivamente el Proyecto Genoma Humano calculándose
quince años de trabajo. Sus objetivos principales en una primera etapa
eran la elaboración de mapas genéticos y físicos de gran resolución,
mientras se ponían a punto nuevas técnicas de secuenciación, para poder
abordar todo el genoma. Se calculó que el Proyecto Genoma Humano
estadounidense necesitaría unos 3000 millones de dólares y terminaría en
2005. En 1993 los fondos públicos aportaron 170 millones de dólares,
mientras que la industria gastó aproximadamente 80 millones. Con el paso
de los años, la inversión privada cobró relevancia y amenazó con
adelantar a las financiaciones públicas.
El proyecto
genoma humano tiene una extensión que es el proyecto microbioma humano .
El mismo intenta caracterizar las comunidades microbianas encontradas
en diversas localizaciones del cuerpo humano para determinar las
posibles correlaciones entre los cambios del microbioma y el estado de
salud.
Se consideraría al microbioma como la rama más alta de al último órgano humano por investigar.
Componentes
Cromosomas
El genoma humano (como el de cualquier organismo eucariota) está formado por cromosomas, que son largas secuencias continuas de ADN altamente organizadas espacialmente (con ayuda de proteínas histónicas y no histónicas) para adoptar una forma ultracondensada en metafase. Son observables con microscopía óptica convencional o de fluorescencia mediante técnicas de citogenética y se ordenan formando un cariotipo.
El cariotipo humano normal contiene un total de 23 pares de cromosomas distintos: 22 pares de autosomas
más 1 par de cromosomas sexuales que determinan el sexo del individuo.
Los cromosomas 1-22 fueron numerados en orden decreciente de tamaño en
base al cariotipo. Sin embargo, posteriormente pudo comprobarse que el
cromosoma 22 es en realidad mayor que el 21.
Las células somáticas de un organismo poseen en su núcleo
un total de 46 cromosomas (23 pares): una dotación de 22 autosomas
procedentes de cada progenitor y un par de cromosomas sexuales, un cromosoma X de la madre y un X o un Y del padre. (Ver imagen 1). Los gametos -óvulos y espermatozoides- poseen una dotación haploide de 23 cromosomas
ADN intragénico
Genes
Un gen es la unidad básica de la herencia, y porta la información genética necesaria para la síntesis de una proteína (genes codificantes) o de un ARN no codificante (genes de ARN). Está formado por una secuencia promotora, que regula su expresión, y una secuencia que se transcribe, compuesta a su vez por: secuencias UTR (regiones flanqueantes no traducidas), necesarias para la traducción
y la estabilidad del ARNm, exones (codificantes) e intrones, que son
secuencias de ADN no traducidas situadas entre dos exones que serán
eliminadas en el procesamiento del ARNm (ayuste).
Actualmente se estima que el genoma humano contiene entre 20 000 y 25 000 genes
codificantes de proteínas, estimación muy inferior a las predicciones
iniciales que hablaban de unos 100 000 genes o más. Esto implica que el
genoma humano tiene menos del doble de genes que organismos eucariotas mucho más simples, como la mosca de la fruta o el nematodoCaenorhabditis elegans. Sin embargo, las células humanas recurren ampliamente al splicing (ayuste) alternativo para producir varias proteínas distintas a partir de un mismo gen, como consecuencia de lo cual el proteoma humano es más amplio que el de otros organismos mucho más simples. En la práctica, el genoma tan sólo
porta la información necesaria para una expresión perfectamente
coordinada y regulada del conjunto de proteínas que conforman el
proteoma, siendo éste el encargado de ejecutar la mayor parte de las
funciones celulares.
Con base en los resultados iniciales arrojados por el proyecto ENCODE (acrónimo de ENCyclopedia Of DNA Elements),
algunos autores han propuesto redefinir el concepto actual de gen. Las
observaciones más recientes hacen difícilmente sostenible la visión
tradicional de un gen, como una secuencia formada por las regiones UTRs,
los exones y los intrones. Estudios detallados han hallado un número de
secuencias de inicio de transcripción por gen muy superior a las
estimaciones iniciales, y algunas de estas secuencias se sitúan en
regiones muy alejadas de la traducida, por lo que los UTR 5' pueden
abarcar secuencias largas dificultando la delimitación del gen. Por otro
lado, un mismo transcrito puede dar lugar a ARN maduros totalmente
diferentes (ausencia total de solapamiento), debido a una gran
utilización del splicing alternativo. De este modo, un mismo
transcrito primario puede dar lugar a proteínas de secuencia y
funcionalidad muy dispar. En consecuencia, algunos autores han propuesto
una nueva definición de gen,:la unión de secuencias genómicas que codifican un conjunto coherente de productos funcionales, potencialmente solapantes.
De este modo, se identifican como genes los genes ARN y los conjuntos
de secuencias traducidas parcialmente solapantes (se excluyen, así, las
secuencias UTR y los intrones, que pasan a ser considerados como
"regiones asociadas a genes", junto con los promotores). De acuerdo con
esta definición, un mismo transcrito primario que da lugar a dos
transcritos secundarios (y dos proteínas) no solapantes debe
considerarse en realidad dos genes diferentes, independientemente de que
estos presenten un solapamiento total o parcial de sus transcritos
primarios.
Las nuevas evidencias aportadas por ENCODE, según las cuales las
regiones UTR no son fácilmente delimitables y se extienden largas
distancias, obligarían a reidentificar nuevamente los genes que en
realidad componen el genoma humano. De acuerdo con la definición
tradicional (actualmente vigente), sería necesario identificar como un
mismo gen a todos aquellos que muestren un solapamiento parcial
(incluyendo las regiones UTR y los intrones), con lo que a la luz de las
nuevas observaciones, los genes incluirían múltiples proteínas de
secuencia y funcionalidad muy diversa. Colateralmente se reduciría el
número de genes que componen el genoma humano. La definición propuesta,
en cambio, se fundamenta en el producto funcional del gen, por lo que se
mantiene una relación más coherente entre un gen y una función
biológica. Como consecuencia, con la adopción de esta nueva definición,
el número de genes del genoma humano aumentará significativamente.
Genes de ARN
Además de los genes codificantes de proteínas, el genoma humano contiene varios miles de genes ARN, cuya transcripción reproduce ARN de transferencia (ARNt), ARN ribosómico (ARNr), microARN (miARN), u otros genes ARN no codificantes. Los ARN ribosómico y de transferencia son esenciales en la constitución de los ribosomas y en la traducción
de las proteínas. Por su parte, los microARN tienen gran importancia en
la regulación de la expresión génica, estimándose que hasta un 20-30 %
de los genes del genoma humano puede estar regulado por el mecanismo de
interferencia por miARN. Hasta el momento se han identificado más de 300
genes de miARN y se estima que pueden existir unos 500.
Distribución de genes
A continuación se muestran algunos valores promedio del genoma
humano. Cabe advertir, sin embargo, que la enorme heterogeneidad que
presentan estas variables hace poco representativos a los valores
promedio, aunque tienen valor orientativo.
La densidad media de genes es de 1 gen cada 100 kb, con un tamaño
medio de 20-30 kb, y un número de exones promedio de 7-8 por cada gen,
con un tamaño medio de 150 nucleótidos. El tamaño medio de un ARNm es de
1.8-2.2 kb, incluyendo las regiones UTR (regiones no traducidas flanqueantes), siendo la longitud media de la región codificante de 1.4 kb.
Variación estructural
Este tipo de variaciones se refiere a duplicaciones, inversiones,
inserciones o variantes en el número de copias de segmentos grandes del
genoma (por lo general de 1000 nucléotidos o más). Estas variantes
implican a una gran proporción del genoma, por lo que se piensa que son,
al menos, tan importantes como los SNPs
Variación estructural es el término general para abarcar un grupo de
alteraciones genómicas que implican segmentos de ADN mayores de 1 Kb. La
variación estructural puede ser cuantitativa (variante en número de
copia, que comprende: deleciones, inserciones y duplicaciones),
posicional (translocaciones) y orientacional (inversiones).
A pesar de que este campo de estudio es relativamente nuevo (los
primeros estudios a gran escala se publicaron en los años 2004 y 2005),
ha tenido un gran auge, hasta el punto de que se ha creado un nuevo
proyecto para estudiar este tipo de variantes en los mismos individuos
en los que se basó el Proyecto HapMap.
Aunque aún quedan dudas acerca de las causas de este tipo de
variantes, cada vez existe más evidencia a favor de que es un fenómeno
recurrente que todavía continua moldeando y creando nuevas variantes del
genoma.
Este tipo de variaciones han potenciado la idea de que el genoma
humano no es una entidad estática, sino que se encuentra en constante
cambio y evolución.
Enfermedades genéticas
La alteración de la secuencia de ADN que constituye el genoma humano
puede causar la expresión anormal de uno o más genes, originando un
fenotipo patológico. Las enfermedades genéticas pueden estar causadas
por mutación de la secuencia de ADN, con afectación de la secuencia codificante (produciendo proteínas incorrectas)
o de secuencias reguladoras (alterando el nivel de expresión de un
gen), o por alteraciones cromosómicas, numéricas o estructurales. La
alteración del genoma de las células germinales de un individuo se
transmite frecuentemente a su descendencia. Actualmente el número de
enfermedades genéticas conocidas es aproximadamente de 4 000, siendo la
más común la fibrosis quística.
El estudio de las enfermedades genéticas frecuentemente se ha
englobado dentro de la genética de poblaciones. Los resultados del
Proyecto Genoma Humano son de gran importancia para la identificación de
nuevas enfermedades genéticas y para el desarrollo de nuevos y mejores
sistemas de diagnóstico genético, así como para la investigación en
nuevos tratamientos, incluida la terapia génica.
Mutaciones
Las mutaciones génicas pueden ser:
Sustituciones (cambios de un nucleótido por otro): Las
sustituciones se denominan transiciones si suponen un cambio entre bases
del mismo tipo químico, o transversiones si son un cambio purina (A, G)→pirimidina (C, T) o pirimidina→purina.
Deleciones o inserciones: son respectivamente la eliminación o
adición de una determinada secuencia de nucleótidos, de longitud
variable. Las grandes deleciones pueden afectar incluso a varios genes,
hasta el punto de ser apreciables a nivel cromosómico con técnicas de
citogenética. Inserciones o deleciones de unas pocas pares de bases en
una secuencia codificante pueden provocar desplazamiento del marco de lectura (frameshift), de modo que la secuencia de nucleótidos del ARNm se lee de manera incorrecta.
Las mutaciones génica pueden afectar a:
ADN codificante: Si el cambio en un nucleótido provoca en
cambio de un aminoácido de la proteína la mutación se denomina no
sinónima. En caso contrario se denominan sinónimas o silenciosas
(posible porque el código genético es degenerado). Las mutaciones no sinónimas asimismo se clasifican en mutaciones con cambio de sentido (missense) si provocan el cambio de un aminoácido por otro, mutaciones sin sentido (non-sense) si cambian un codón codificante por un codón de parada (TAA, TAG, TGA) o con ganancia de sentido si sucede a la inversa.
ADN no codificante: Pueden afectar a secuencias reguladoras,
promotoras o implicadas en el ayuste (splicing). Estas últimas pueden
causar un erróneo procesamiento del ARNm, con consecuencias diversas en
la expresión de la proteína codificada por ese gen.
Trastornos monogénicos
Son enfermedades genéticas causadas por mutación en un sólo gen, que
presentan una herencia de tipo mendeliano, fácilmente predecible. En la
tabla se resumen los principales patrones de herencia que pueden
mostrar, sus características y algunos ejemplos.
Patrón hereditario
Descripción
Ejemplos
Autosómico dominante
Enfermedades que se manifiestan en individuos heterocigóticos.
Es suficiente con una mutación en una de las dos copias (recuérdese que
cada individuo posee un par de cada cromosoma) de un gen para que se
manifieste la enfermedad. Los individuos enfermos generalmente tienen
uno de sus dos progenitores enfermos. La probabilidad de tener
descendencia afectada es del 50 % dado que cada progenitor aporta uno de
los cromosomas de cada par. Frecuentemente corresponden a mutaciones
con ganancia de función (de modo que el alelo mutado no es inactivo sino
que posee una nueva función que provoca el desarrollo de la enfermedad)
o por pérdida de función del alelo mutado con efecto de dosis génica
también conocido como haploinsuficiencia. Frecuentemente son
enfermedades con baja penetrancia, es decir, sólo una parte de los individuos que portan la mutación desarrollan la enfermedad.
La enfermedad sólo se manifiesta en individuos homocigóticos
recesivos, es decir, aquellos en los que ambas copias de un gen están
mutadas. Son mutaciones que causan pérdida de función, de modo que la
causa de la enfermedad es la ausencia de la acción de un gen. La
mutación sólo en una de las dos copias es compensada por la existencia
de la otra (cuando una sola copia no es suficiente se origina
haploinsuficiencia, con herencia autosómica dominante). Habitualmente un
individuo enfermo tiene ambos progenitores sanos pero portadores de la
mutación (genotipoheterocigótico: Aa). En tal caso un 25 % de la descendencia estará afectada.
Las enfermedades dominantes ligadas al cromosoma X están causadas
por mutaciones en dicho cromosoma, y presentan un patrón hereditario
especial. Sólo unas pocas enfermedades hereditarias presentan este
patrón. Las mujeres tienen mayor prevalencia de la enfermedad que los
hombres, dado que reciben un cromosoma X de su madre y otro de su padre,
cualquiera de los cuales puede portar la mutación. Los varones en
cambio siempre reciben el cromosoma Y de su padre. Así, un varón enfermo
(xY) tendrá todos sus hijos varones sanos (XY) y todas las hijas
enfermas (Xx), mientras que una mujer enferma (Xx) tendrá un 50 % de su
descendencia enferma, independientemente del sexo. Algunas de estas
enfermedades son letales en varones (xY), de modo que sólo existen
mujeres enfermas (y varones con síndrome de Klinefelter, XxY).
Las enfermedades recesivas ligadas al X también están causadas por
mutaciones en el cromosoma X. Los varones están más frecuentemente
afectados. Un varón portador siempre será enfermo (xY) dado que sólo
posee un cromosoma X, que está mutado. Su descendencia serán varones
sanos (XY) e hijas portadoras (Xx). Una mujer portadora, tendrá una descendencia compuesta por un 50 % de hijas portadoras y un 50 % de varones enfermos.
Son enfermedades causadas por mutación en el cromosoma Y. En
consecuencia, sólo puede manifestarse en varones, cuya descendencia será
del 100 % de hijas sanas y el 100 % de hijos varones enfermos. Dadas
las funciones del cromosoma Y, frecuentemente estas enfermedades sólo
causan infertilidad, que a menudo puede ser superada terapéuticamente.
Infertilidad masculina hereditaria
Mitocondrial
Enfermedades causadas por mutación en genes del genoma mitocondrial.
Dadas la particularidades de dicho genoma, su transmisión es
matrilineal (el genoma mitocondrial se transfiere de madres a hijos). La
gravedad de una mutación depende del porcentaje de genomas afectados en
la población de mitocondrias, fenómeno denominado heteroplasmia (en
contraste con heterocigosis), que varía por segregación mitótica
asimétrica.
Neuropatía óptica hereditaria de Leber (LHON)
Trastornos poligénicos y multifactoriales
Otras alteraciones genéticas pueden ser mucho más complejas en su
asociación con un fenotipo patológico. Son las enfermedades
multifactoriales o poligénicas, es decir, aquellas que están causadas
por la combinación de múltiples alelos genotípicos y de factores
exógenos, tales como el ambiente o el estilo de vida. En consecuencia no
presentan un patrón hereditario claro, y la diversidad de factores etiológicos y de riesgo dificulta la estimación del riesgo, el diagnóstico y el tratamiento.
Algunos ejemplos de enfermedades multifactoriales con etiología parcialmente genética son:
autismo
enfermedad cardiovascular
hipertensión
diabetes
obesidad
cáncer
Alteraciones cromosómicas
Las alteraciones genéticas pueden producirse también a escala cromosómica (cromosomopatías), causando severos trastornos que afectan a múltiples genes y que en muchas ocasiones son letales provocando abortos prematuros. Frecuentemente están provocadas por un error durante la división celular,
que sin embargo no impide su conclusión. Las alteraciones cromosómicas
reflejan una anormalidad en el número o en la estructura de los
cromosomas, por lo que se clasifican en numéricas y estructurales.
Provocan fenotipos muy diversos, pero frecuentemente presentan unos
rasgos comunes:
Retraso mental y retraso del desarrollo.
Alteraciones faciales y anomalías en cabeza y cuello.
Malformaciones congénitas, con afectación preferente de extremidades, corazón, etc.
Numéricas
Frecuencias de aneuploidías por cada 1000 nacidos vivos.15
Aneuploidía
Frecuencia
(/1000)
Síndrome
Trisomía 21
1.5
de Down
Trisomía 18
0.12
de Edwards
Trisomía 13
0.07
de Patau
Monosomía X
0.4
de Turner
XXY
1.5
de Klinefelter
XYY
1.5
del XYY
Es una alteración del número normal de cromosomas de un individuo,
que normalmente presenta 23 pares de cromosomas (46 en total), siendo
cada dotación cromosómica de un progenitor (diploidía). Si la alteración afecta a un sólo par de cromosomas se habla de aneuploidía, de manera que puede haber un sólo cromosoma (monosomía) o más de dos (trisomía, tetrasomía...). Un ejemplo de gran prevalencia
es la trisomía 21, responsable del Síndrome de Down. Si por el
contrario la alteración afecta a todos los cromosomas se habla de euploidías, de manera que en teoría el individuo tiene una sola dotación cromosómica (haploidía, 23 cromosomas en total) o más de dos dotaciones (triploidía: 69 cromosomas; tetraploidía:
92 cromosomas...). En la práctica las euploidías causan letalidad
embronaria (abortos) siendo muy pocos los nacidos vivos, y fallecen muy
tempranamente. Las aneuploidías son mayoritariamente letales, salvo las
trisomías de los cromosomas 13, 18, 21, X e Y (XXY, XYY), y la monosomía
del cromosoma X. En la tabla se muestran las frecuencias de nacidos
vivos con estas alteraciones.
Estructurales
Se denominan así las alteraciones en la estructura de los cromosomas,
tales como las grandes deleciones o inserciones, reordenaciones del
material genético entre cromosomas... detectables mediante técnicas de
citogenética.
Deleciones: eliminación de una porción del genoma. Algunos trastornos conocidos son el síndrome de Wolf-Hirschhorn por deleción parcial del brazo corto del cromosoma 4 (4p), y el síndrome de Jacobsen o deleción 11q terminal.
Duplicaciones: una región considerable de un cromosoma se duplica. Un ejemplo es la enfermedad de Charcot-Marie-Tooth
tipo 1A, que puede ser causada por duplicación del gen codificante de
la proteína mielínica periférica 22 (PMP22) en el cromosoma 17.
Translocaciones:
cuando una porción de un cromosoma se transfiere a otro cromosoma. Hay
dos tipos principales de translocaciones: la translocación recíproca, en
la que se intercambian segmentos de dos cromosomas distintos, y la
translocación Robertsoniana, en la que dos cromosomas acrocéntricos (13, 14, 15, 21, 22) se fusionan por sus centrómeros (fusión céntrica).
Inversiones:
una parte del genoma se rompe y se reorienta en dirección opuesta antes
de reasociarse, con lo que dicha secuencia aparece invertida. Pueden
ser paracéntricas (si afectan sólo a una brazo) o pericéntricas (si la
secuencia invertida incluye el centrómero).
Cromosomas en anillos: una porción del genoma se rompe y forma un
anillo por circularización. Esto puede ocurrir con pérdida de material o
sin pérdida de material.
Isocromosomas:
cromosomas simétricos, con sus dos brazo idénticos por deleción de uno
de los brazos y duplicación del otro. El más habitual es el isocromosoma
X, en el que se pierde el brazo corto del cromosoma X, originando
fenotipos de Síndrome de Turner.
Los síndromes de inestabilidad cromosómica son un grupo de trastornos
caracterizados por una gran inestabilidad de los cromosomas, que sufren
con gran frecuencia alteraciones estructurales. Están asociados con un
aumento de la malignidad de neoplasias.
Evolución
Los estudios de genómica comparada se basan en comparación de
secuencias genómicas a gran escala, generalmente mediante herramientas bioinformáticas.
Dichos estudios permiten ahondar en el conocimiento de aspectos
evolutivos de escala temporal y espacial muy diversa, desde el estudio
de la evolución de los primeros seres vivos hace miles de millones de
años o las radiaciones filogenéticas en mamíferos, hasta el estudio de
las migraciones de seres humanos en los últimos 100 000 años, que
explican la actual distribución de las distintas razas humanas.
Genómica comparada entre distintas especies
Los estudios de genómica comparada con genomas de mamíferos sugieren
que aproximadamente el 5 % del genoma humano se ha conservado
evolutivamente en los últimos 200 millones de años; lo cual incluye la
gran mayoría de los genes y secuencias reguladoras. Sin embargo, los
genes y las secuencias reguladoras actualmente conocidas suponen sólo el
2 % del genoma, lo que sugiere que la mayor parte de la secuencia
genómica con gran importancia funcional es desconocida. Un porcentaje
importante de los genes humanos presenta un alto grado de conservación
evolutiva. La similitud entre el genoma humano y el del chimpancé (Pan troglodytes) es del 98.77 %. En promedio, una proteína humana se diferencia de su ortóloga de chimpancé en tan sólo dos aminoácidos,
y casi un tercio de los genes tiene la misma secuencia. Una diferencia
importante entre los dos genomas es el cromosoma 2 humano, que es el
producto de una fusión entre los cromosomas 12 y 13 del chimpancé
Otra conclusión de la comparación del genoma de distintos primates es
la notable pérdida de genes de receptores olfativos que se ha producido
paralelamente al desarrollo de la visión en color (tricrómica) durante
la evolución de primates.
Crean un catálogo universal de los errores genéticos del ser humano
Las
enfermedades hereditarias se producen porque algunos genes del
organismo acumulan errores, las llamadas mutaciones. Estos genes son
secuencias de ADN que contienen las instrucciones para que el cuerpo
funcione, crezca y se reproduzca pero, cuando aparecen estos fallos, las
instrucciones dejan de ser correctas y la maquinaria se estropea. Para
conseguir que el «motor» vuelva a girar, puede ser muy interesante tener
un registro con todas las posibles averías. Del mismo modo, si los
científicos tuvieran a su alcance un catálogo de todos los posibles
errores genéticos, con la frecuencia y el lugar donde aparecen, y si
pudieran relacionarlos con las enfermedades que provocan, la medicina
daría un gran salto al futuro: sería enormemente más fácil investigar y
curar enfermedades hereditarias
Esta
es la filosofía que ha llevado este miércoles a la publicación de un
artículo en la revista «Nature» en el que se ha presentado el que es,
hasta ahora, el catálogo más completo de las mutaciones humanas. Gracias
al trabajo de decenas de investigadores de todo el mundo, a lo largo de
14 estudios unidos en el Consorcio de Agregación de Exomas (ExAC), los
científicos han secuenciado genes de 60.706 personas, una cifra que
multiplica por diez al número de individuos analizados en previos
estudios. Gracias a esto, se han identificado un total de 7,5 millones
de mutaciones, lo que facilitará la investigación y futuro tratamiento
de las dolencias hereditarias, en especial si se tratan de enfermedades
raras.
«El avance más importante es que se ha puesto a disposición
de la comunidad médica un catálogo de mutaciones genéticas de la
especie humana, con su localización y su frecuencia», ha explicado a ABC
Roberto Elosua, investigador del Instituto Hospital del Mar de
Investigaciones Médicas (IMIM) y parte del equipo de científicos
responsable de este proyecto.
En un esfuerzo científico y
político sin precedentes, investigadores de todo el mundo han cooperado
para compartir gratuitamente la información recogida con el resto del
mundo: «Esta investigación ya está siendo muy importante para que la
comunidad médica estudie las enfermedades hereditarias. Para eso es
crucial entender que esa información hay que compartirla y que no es de
nadie, que es patrimonio de la humanidad. Solo así toda la población
puede beneficiarse», ha señalado Elosua.
Esta iniciativa permitirá
facilitar la investigación y el diagnóstico de las cerca de 4.700
deficiencias hereditarias que se conocen y que dependen de un solo gen,
entre las que están la enfermedad de Huntington, la distrofia muscular
de Duchenne, la hemofilia A, las hipercolesterolemias familiares, las
miocardiopatías de base genética o algunos síndromes de muerte súbita.
Genética universal
Además,
gracias a la gran cantidad de individuos analizados, 60.706, este
catálogo ha logrado reunir una base de datos universal de la
variabilidad genética del ser humano. La información recogida recopila
las mutaciones típicas de personas de origen africano, asiático y
latino, para incluirla a los datos sobre personas de origen caucásico. Aparte
de facilitar la investigación y el diagnóstico de enfermedades
hereditarias, este catálogo ha logrado otro gran éxito. Ha permitido
detectar, entre los 20.000 genes humanos, alrededor de 3.200 que se caracterizan por estar intactos
y no acumular mutaciones. «Están muy conservados, lo que quiere decir
que son muy importantes para la supervivencia o para la reproducción de
la especie», ha señalado Elosua. Esto, tal como ha dicho, abre nuevos
interrogantes sobre la función de estos genes clave.
Un nudo gordiano
Junto a estos genes que apenas varían, esta colosal investigación ha identificado 180.000 nuevas mutaciones
que podrían estar implicadas en enfermedades. Curiosamente, también se
ha descubierto que mutaciones normalmente asociados a enfermedades en
realidad no lo están. Todos estos avances en la genética de las
enfermedades es solo una muestra de lo que una secuenciación tan
exhaustiva del genoma puede lograr en el futuro. Pero relacionar
mutaciones y dolencias no es entender la base genética de las
enfermedades. Al igual que ocurre con la neurología o la física de
partículas, destinadas a enfrentarse a los profundos misterios del
cerebro y de los átomos, los genetistas se enfrentan a una abrumadora complejidad en el mundo de los genes. En este sentido, Jay Shendure
investigador en la Universidad de Washington, ha escrito, en un
artículo que ha acompañado en «Nature» al estudio de los 60.706 genomas,
que este exhaustivo catálogo de mutaciones apenas «proporciona un primer vistazo de la variabilidad genética en humanos».
Elosua ha coincidido en este sentido, al señalar que «de momento, lo
que estamos entendiendo es el código de las secuencias». Una norma que
depende de unas letras que forman los genes, pero a la que hay que sumar
el modo como se colocan y estructuran y, por último, la manera como se
regula su lectura, a través de la llamada epigenética. «Hay
diferentes capas de complejidad y de lenguajes que hacen que la
dificultad de comprender la función de los genes sea extraordinariamente
alta»,
en palabras de Elosua. Por eso, más que nunca, la colaboración y la
inversión en grandes proyectos de secuenciación promete ampliar
enormemente las fronteras del conocimiento, y borrar el adjetivo
«incurable» del repertorio de enfermedades hereditarias.